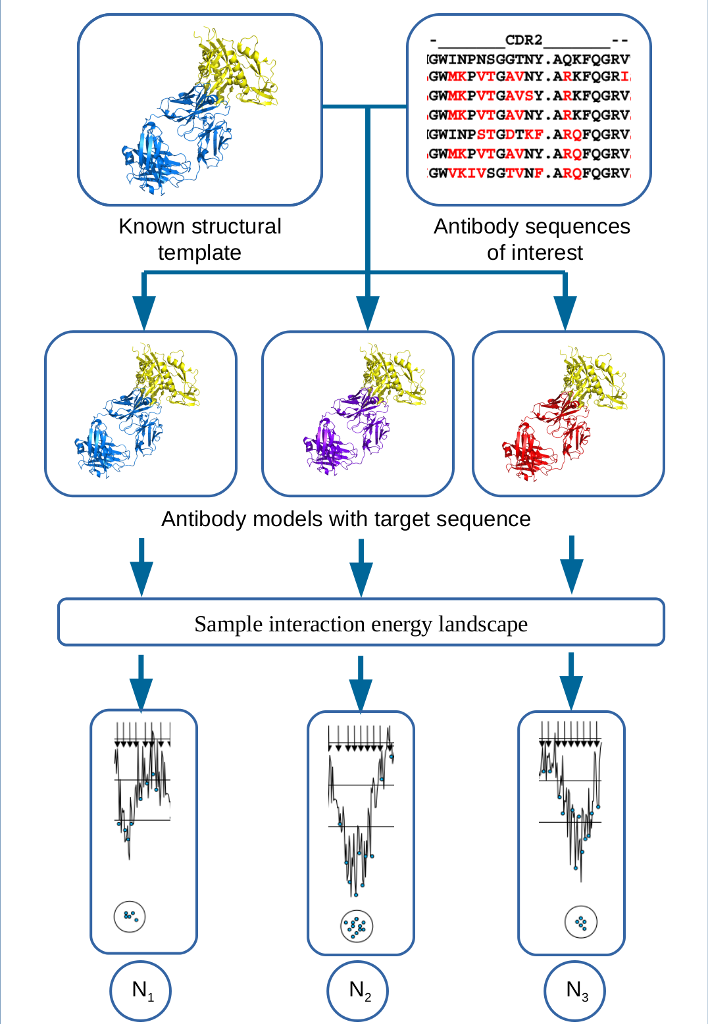
The ability to predict the changes in binding affinity due to amino acid mutations is important for better understanding of the process of affinity maturation. It is well known that the prediction of binding free energy remains generally problematic. The detailed thermodynamic simulations are too time consuming, whereas the empirical and semi-empirical approaches may not be accurate enough. Thus the need for new approaches to binding free energy calculation is generally recognized, and any advance in methodology will have substantial impact. In addition, while modeling has contributed to the design of therapeutic antibodies, notably in early attempts at humanizing antibodies, overall the potential impact of computational methods is not as well defined, and the tools are not as well developed and less broadly employed, than in small molecule drug discovery. There are two main reasons for this difference. First, a crystal structure is frequently not available for most newly developed antibody sequences, in which case a high-resolution antibody homology model is needed to perform structure-based in silico antibody engineering. Docking and calculation of binding free energy changes based on homology models are generally much less reliable than using X-ray structures. Second, antibody maturation may affect both the antigen-antibody interaction energy and the entropy change due to stabilization of hypervariable loops , primarily due to long-range electrostatic interactions of residues that frequently are outside of the interface, which makes the modeling more difficult.