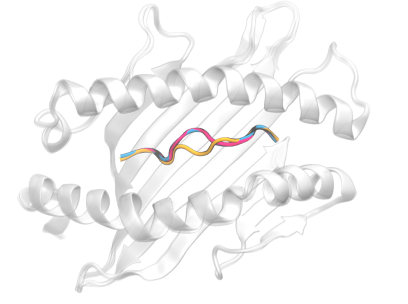
The precise prediction of major histocompatibility complex (MHC)-peptide complex structures is pivotal for understanding cellular immune responses and advancing vaccine design. In our latest study, published in Biophysical Journal, we have enhanced AlphaFold’s capabilities by fine-tuning it with a specialized dataset consisting exclusively of high-resolution class I MHC-peptide crystal structures.
AlphaFold, while broadly effective, lacked the granularity necessary for the high-precision demands of class I MHC-peptide interaction prediction. Our tailored approach addresses this by providing a more detailed and accurate model. A comparative analysis was conducted against the homology-modeling-based method Pandora, as well as the AlphaFold multimer model. Our fine-tuned model demonstrates superior performance, with a median root-mean-square deviation (RMSD) for Cα atoms in peptides of 0.66 Å and improved predicted local distance difference test scores.
Moreover, our additional comparisons with AlphaFold3 on new MHC-I structures from the Protein Data Bank (PDB) published after January 1, 2023, show that our model has 15% more samples under 1 Å deviation, highlighting its enhanced precision.
These advances have substantial implications for computational immunology, potentially accelerating the development of novel therapeutics and vaccines by providing a more precise computational lens through which to view MHC-peptide interactions.
