The team lead by professors Dima Kozakov (Stony Brook University) and Sandor Vajda (Boston University) made remarkable progress in improving the prediction of protein multimers and protein-ligand complexes
Background of CASP
CASP (Critical Assessment of Structure Prediction) is a worldwide experiment for protein structure prediction taking place every two years since 1994. In 2018 CASP has been integrated with CAPRI (Critical Assessment of Predicted Interactions), a similar contest for predicting molecular interactions. In both events participants are expected to predict the structures of proteins or protein complexes that, at the time of the competition, are not yet released. Independent assessors compare the models to experimental structures, The Google-owned company DeepMind participated in CASP-14 in 2020 with their machine learning based protein structure prediction program AlphaFold-2. For many of the protein targets the program predicted structures that were indistinguishable from the structures determined by costly X-ray crystallography or nuclear magnetic resonance experiments. The AlphaFold-2 program, released in in 2021, has been adopted and now is widely used by the biomedical research community. Most participants of the CASP-15 competition in 2022 used modified versions of AlphaFold-2, achieving moderate improvements in the accuracy of the models. The leaders of the AlphaFoild-2 project, Dennis Hassabis and John Jumper, shared the 2024 Nobel prize in chemistry. During the last two years, Google DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI) have partnered to create AlphaFold DB to make the predictions freely available to the scientific community. The latest database release contains over 200 million entries, providing broad coverage of proteins and protein complexes. More recently, DeepMind and its subsidiary, Isomorphic Labs, released the program AlphaFold-3. The new program further increases the accuracy of predicting the structures of proteins and protein complexes, and also enables determining interactions between proteins and small druglike molecules that was not possible using AlphaFold-2.
The most recent CASP contest has been conducted during the summer months of 2024, and results were presented at the CASP16 conference held in Punta Cana, Dominican Republic, on December 1-4, 2024. Based on the evaluator’s report, the team lead by professors Dima Kozakov (Stony Brook University) and Sandor Vajda (Boston University) made remarkable progress in improving the prediction of protein multimers and protein-ligand complexes.
Protein Multimer
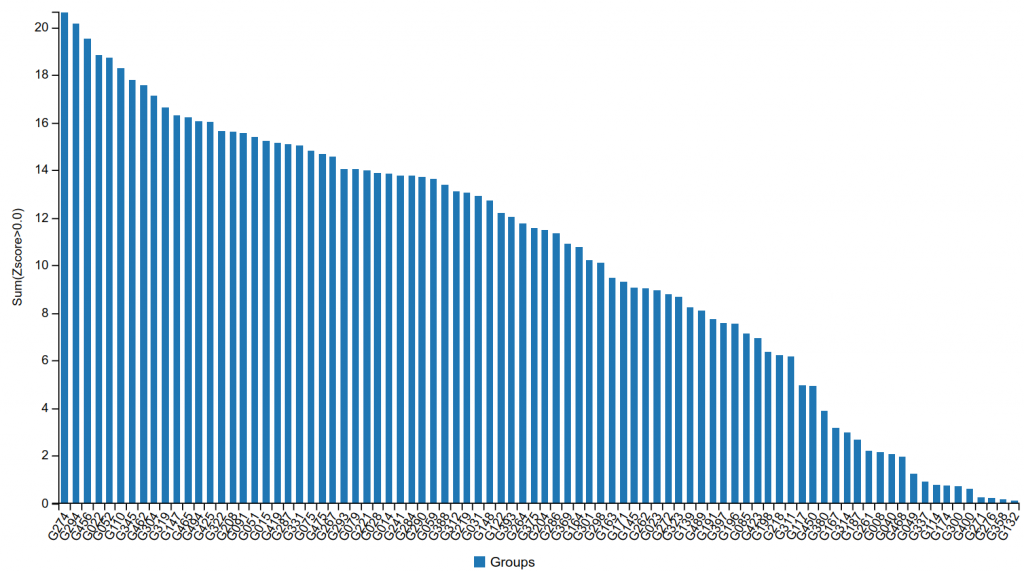
The group submitted models that exceeded the accuracy reached by other participants by a large margin. Figure 1 shows the performance of the participating CASP+CAPRI groups on CAPRI targets, with G274 being the identification number of the Kozakov/Vajda group. As shown, the group substantially outperformed all other teams, in spite of the fact that all groups had access to the latest versions of the AlphaFold-2 and the AlphaFold-3 programs, and the methods used by most groups incorporated these tools in some form.
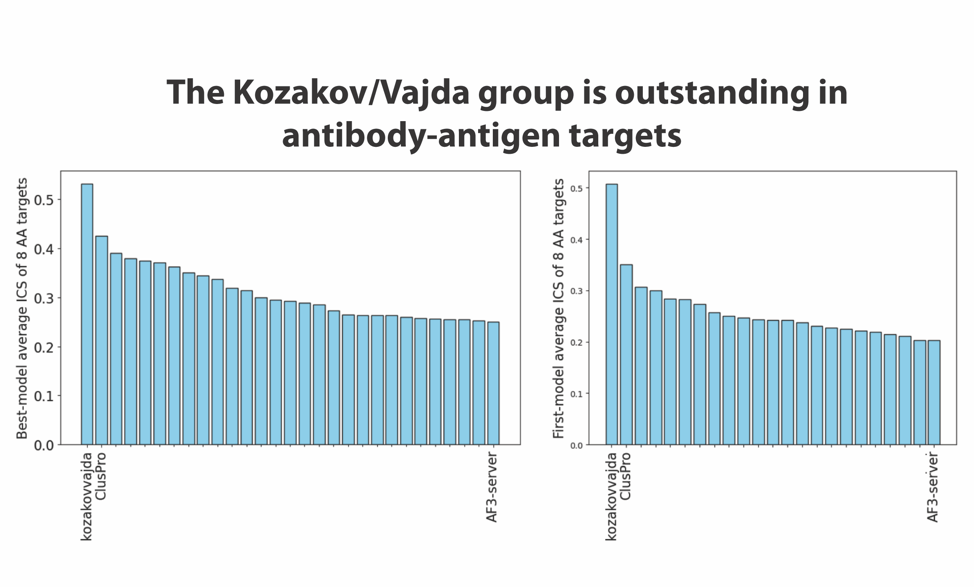
We note that this increase in accuracy was possible because the targets of CASP-16 included several antibody-antigen complexes, and both AlphaFold-2 and AlphaFold-3 are known to perform relatively poorly when predicting the structures of such multimers. In fact, as shown in Figure 2 released by the CASP assessors, the Kozakov/Vajda team obtained very good results for such complexes. It may be interesting to note that their results are much better than the ones produced by AlphaFold-3, which already made improvements relative to the AlphaFold-2 models of antibody-antigen complexes. The results displayed in Figure 2 were presented by the assessors at the CASP-16 meeting. ClusPro is the prediction server of the Kozakov/Vajda group. They participated both as a human predictor group and as a server, which is allowed by the CASP/CAPRI rules, and both teams obtained very good results, with ClusPro being the second-best predictor.
Protein-Ligand
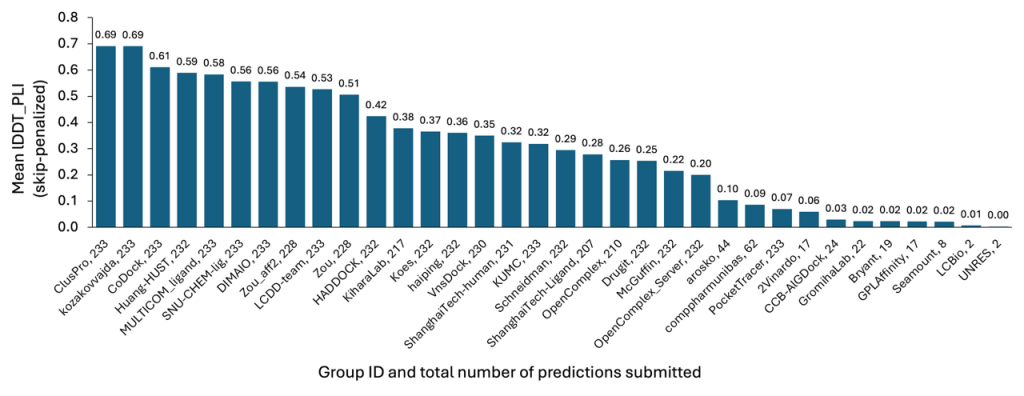
As with the Protein Multimer section, models submitted by our team attained the highest accuracy among all participants, as presented in Figure 3. This achievement stems from our efficient methods, which allowed for the rapid and economical exploration of target conformational space and the subsequent identification of the most favorable poses for submission.
Conclusion
The relatively high prediction accuracy by the Kozakov/Vajda team is due to the unique protocol developed by the two labs during the last three years. The central idea of the approach is integrating the physics of protein interactions and the geometry of the conformational space into the machine learning models. In the current machine learning models, the sampling schedule tends to be biased by the training data. Thus, when required to predict novel interactions not encountered in the training, sampling becomes essentially random, which makes the method very inefficient due to the vast conformational space. In contrast, the Kozakov/Vajda team employs an ML method that systematically samples regions of interest, allowing the identification of correct structures in a rational and efficient manner. This systematic sampling is enabled by fast Fourier transform (FFT)-based methods of evaluating the energies of docked structures. The approach is being implemented in the widely used ClusPro server for predicting the structure of macromolecular interactions, which currently has nearly 40,000 users. While the method demonstrated success at CASP in specific tasks, the core principle of combining machine learning with physics-based sampling is expected to enhance performance in a variety of applications, particularly when the available data are insufficient for effective training.