“On mathematical aspects of molecular replacement”, after G. S. Chirikjian et al
Sergiy Merenkov (CUNY City College of New York)
Kozakov lab
“On mathematical aspects of molecular replacement”, after G. S. Chirikjian et al
Sergiy Merenkov (CUNY City College of New York)
“Generalized Born models”
Maria Sabitova (CUNY Queens College)
A series of talks on “Fast calculation of Ewald sum for mutual orientation of two rigid bodies within periodic cell, using generalized Fourier transforms”, Part III
Maria Sabitova (CUNY Queens College)
A series of talks on “Fast calculation of Ewald sum for mutual orientation of two rigid bodies within periodic cell, using generalized Fourier transforms”, Part II
Maria Sabitova (CUNY Queens College)
A series of talks on “Fast calculation of Ewald sum for mutual orientation of two rigid bodies within periodic cell, using generalized Fourier transforms”, Part I
Maria Sabitova (CUNY Queens College)
We present an ultra-fast approach to modeling protein interactions.
Protein-protein interactions (PPIs) are the basis of cellular functions, and when these processes are compromised diseases such as cancer emerge. For years scientists have tried with mixed success to map out PPIs to understand cellular processes. Now our group has outlined a method that could pave the way to designing new drugs that prevent problematic protein interactions that lead to disease. The findings are published in the early online edition of PNAS.
Proteins are the major building blocks of the cell. Many proteins perform their function by interacting with other proteins. In a typical cell, hundreds of thousands of different protein interactions take place. Characterizing the structure of these interactions helps elucidate how organisms function normally and during disease development.
The problem considered is given three dimensional structures of two individual proteins to predict how these protein interact with each other. We can liken the method to characterizing all the possible structures that pairs of “lego blocks” form out of a huge set of different individual starting blocks.
In the paper titled “Protein-protein docking by fast generalized Fourier transforms on 5D rotational manifolds,” we explain a new algorithm used to create ultra-fast approach to modeling protein interactions. We discovered that the method runs an order of magnitude faster than previous state-of-the-art methods and has comparable accuracy.
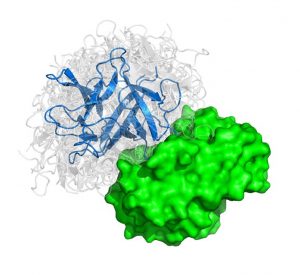
The algorithm features a fast Manifold Fourier transform (FMFT) that helps to speed the calculations, enabling us to sample a large number of putative protein-protein complex conformations.
The new algorithm will soon be available to the scientific community through our publicly available protein-protein docking server called ClusPro. This resource, with more than 15000 academic users worldwide, supported by the National Science Foundation and the Binational Science Foundation, is being developed by our group in collaboration with scientists at Boston University. ClusPro was judged to be the best automated docking server in the latest rounds of the international blind protein docking competition called CAPRI (Critical Assessment of Prediction Interaction).
CAPRI (Critical Assessment of Predicted Interactions) experiment is a community-wide effort dedicated to evaluating the current state of methods for prediction of protein complex structure.
The evaluation of results for the last 7 rounds comprising a total of 20 prediction targets, was recently presented in Tel Aviv at the sixth CAPRI Evaluation Meeting. More then 6000 models submitted by 68 predictor groups and 14 automated servers were processed.
Automated protein docking server ClusPro developed by our own Applied BioComputation group in collaboration with Structural BioInformatics lab at Boston University was ranked first in the server category. The summary of the results is shown below. For each predictor group, the table shows the number of acceptable or better predictions, and among those the number of high quality models, indicated by three stars, as well as the number of medium quality solutions, indicated by two stars.
| Server rankings | |
|---|---|
| Server | Predictions |
| ClusPro | 9/3** |
| PyDockWeb | 6/2** |
| LZerD | 4/1***/1** |
| HADDOCK | 4/2** |
| SwarmDock | 3/2** |
| GalaxyPPDock | 1** |
| PatchDock-FiberDock, DOCK/PIERR, MegaDock, GRAMM-X | 1 |
| SurFit | 0 |
Interestingly, the server’s performance was comparable to that of the best human predictor groups, although the latter had access to all information available in the literature. The results for the 10 best-performing groups are provided below for comparison.
| Human predictor rankings | |
|---|---|
| Group | Predictions |
| Guerois | 10/1***/8** |
| Zacharias | 10/3***/2** |
| Vajda/Kozakov, Seok | 8/3***/2** |
| Weng | 6/1***/4** |
| Fernandez-Recio | 7/1***/3** |
| Vakser | 6/2***/2** |
| Eisenstein | 4/2***/2** |
| Zou | 7/1***/2** |
| Bates | 6/3** |
| Huang | 5/3*** |
We are happy to announce that our lab has won a $50K grant from the Long Island Bioscience Hub (LIBH) for our project titled Fast Hybrid Approach For Determination of Structure of Therapeutic Protein Complexes. LIBH represents an active collaboration between the Center for Biotechnology, Stony Brook University, Cold Spring Harbor Laboratory, Brookhaven National Laboratory, and the Feinstein Institute at Northwell Health System, and its technology development awards aim to speed up the commercialization of novel biomedical technologies and support the formation of new companies both in the region and throughout the New York State.